Methods for interpreting cryo-EM maps
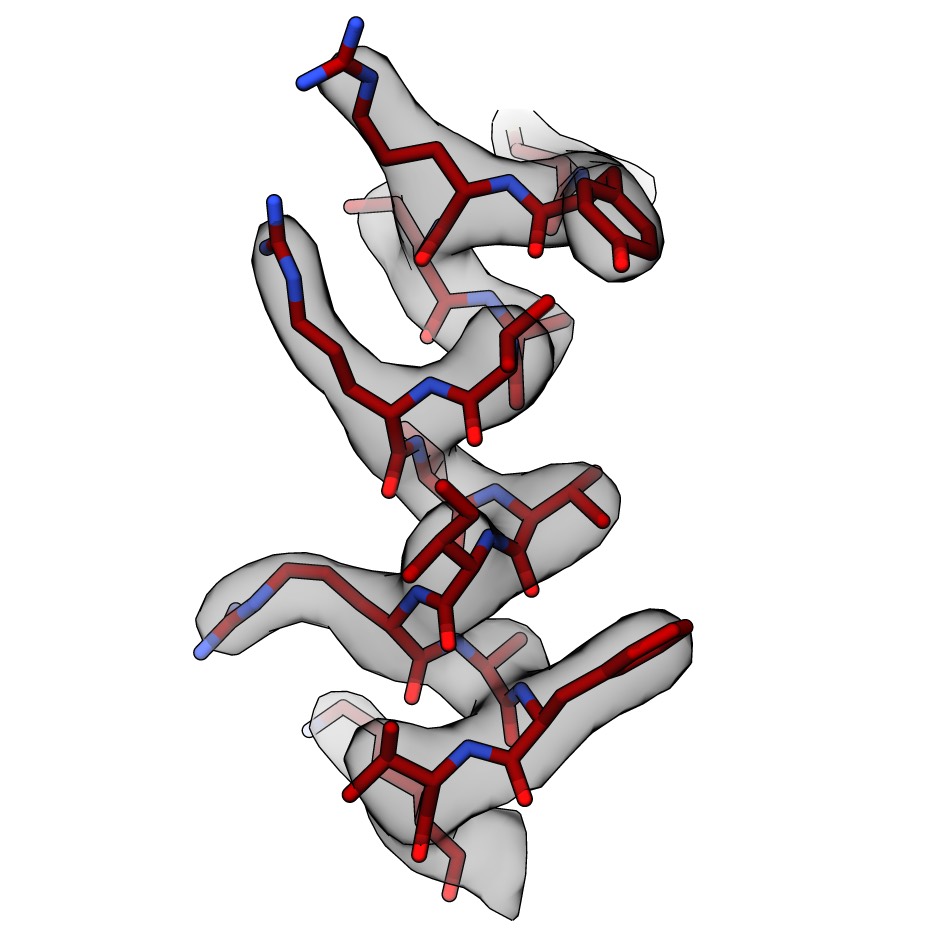
We are also interested in developing new methods to accelerate and improve cryo-EM structure determination. In particular how we can improve the interpretation of cryo-EM density maps with all-atom models and make these models as accurate as possible. Some useful tools that we helped develop for cryo-EM can be found here. More recently, we have worked with Kiarash Jamali and Sjors Scheres at the MRC-LMB to test and extend ModelAngelo, a deep-learning-based approach to automated model building, for de novo identification of proteins from cryo-EM maps. A preprint of this work can be found on BioRxiv and the software downloaded from GitHub.